
Steve Vigdor, April 4, 2019
In Part II of this blog series, we discussed the ample evidence for the roles of mutations and natural selection in microevolution provided by laboratory experiments on the development of bacteria and antibodies. We also presented some of the extensive fossil evidence supporting the concept of universal common descent in macroevolution. In this part, we discuss the compelling biomolecular evidence for common descent that has arisen in the wake of the mid-20th century discovery of the structure of DNA. We discuss the nature of genetic mutations and how they occur during DNA replication, together with the natural selection of beneficial mutations. Then we present some of the striking similarities in DNA and protein sequences among different species that have been revealed in the 21st century by the rapid advancements in mapping of the full genome of multiple species. Parts of the discussion here follow sections in chapter 7 of my book Signatures of the Artist: The Vital Imperfections that Make Our Universe Habitable. That chapter deals with the essential roles played by randomness in the quantum world, in seeding the large-scale structure of the universe, and in the origins and evolution of life.
11) DNA and DNA Coding
All biologically viable systems rely on the storage of genetic information in complex polymer molecules that are capable of being replicated reasonably faithfully in biochemical processes. In the vast majority of living species the relevant biomolecules are DNA (deoxyribonucleic acid). Some simple organisms, including many viruses (e.g., those that cause ebola, influenza, West Nile fever, polio and measles), rely instead on RNA (ribonucleic acid). The physical and chemical structures of RNA and DNA are compared in Fig. 11.1. The double helical structure of DNA, as well as the properties of its sugar-phosphate backbone, give it greater chemical stability than RNA. DNA is thus capable of forming much longer polymer chains and of storing much more genetic information.

When living cells divide, the DNA or RNA molecules get replicated in the daughter cells. The chemical reactions needed in the case of DNA rely on enzyme proteins to catalyze the replication, by a process that will be described in the next section. Because of its single-stranded structure, some RNA molecules, called ribozymes, are capable of catalyzing their own self-replication. In either case, the replication is not perfect: a nitrogenous base at a particular location may be replaced by a different base or deleted in one of the copies (a so-called point mutation), or small sequences of bases may get reversed or inadvertently repeated. Typical error rates in RNA replication are comparable to or larger than 1% per base. In contrast, enzyme-catalyzed DNA replication is intrinsically much more accurate and has proofreading and post-replication mismatch repair mechanisms that lead typically to part per billion or better error rates. The unique pairwise matching of base pairs in DNA implies that each strand in the double helix contains the same genetic information, a feature vital in DNA replication.
Because of its greater chemical stability and replication fidelity, DNA provides a far more reliable genetic storage mechanism to ensure robust reproduction within long-lasting, complex species. On the other hand, the shorter polymer lengths and more error-prone copying of RNA may have served a very useful purpose in allowing more rapid synthesis and random sampling along an initial path on Earth toward the first self-replicating biomolecules. This is why speculation about the origins of life often invokes an RNA World hypothesis. And the higher mutation rates in RNA replication allow some viruses to evolve rapidly enough to withstand the onslaught of human immune response and to complicate the development of highly effective vaccines to combat the diseases they cause. In the cells of complex organisms various RNA molecules play supporting roles in DNA replication: for example, transcribing the genetic information stored in long DNA molecules and catalyzing the formation of peptide bonds binding amino acids together into the proteins needed to support DNA replication.
Biological functions of diverse nature in living organisms are performed by thousands of proteins, which are themselves long polymer molecules assembled from strings of amino acids. The amino acids are simple organic compound molecules comprising mainly hydrogen, carbon, oxygen and nitrogen atoms, as indicated in Fig. 11.2. The side chain labeled R in Fig. 11.2 may contain other elements, and differs in nature among different amino acids. Among about 500 naturally occurring amino acids, only 20 are used in constructing proteins in humans. These 20 amino acids form the basic alphabet of life. The instructions for building proteins by stringing amino acids together are encoded in so-called coding DNA, which comprises about 2% of the human genome (i.e., the full set of genes summed over all 46 human chromosomes).

The coding DNA comprises triplets of adjacent base pairs that instruct RNA molecules about which of the 20 amino acid types to assemble next into the proteins needed for expression of a given gene, as well as where in the DNA chain the instructions for that gene begin and end. As illustrated in Fig. 11.3, there is considerable redundancy in this genetic coding, with several different base pair triplet sequences coding the same amino acid. This redundancy ensures that some particular mutations that may occur within coding DNA during replication have no effect at all on protein sequencing; such mutations are called “silent.” There is an analogous, but even more extensive, redundancy in protein function, as we will discuss below: an enormous number of different amino acid sequences can yield proteins executing the same biological function.

The biological purpose of much of non-coding DNA – the long sequences of base pairs that do not encode the construction of proteins – remains a topic of controversy and ongoing genomic research. Some of the non-coding sections are known to get transcribed into non-coding RNA molecules that serve a variety of regulatory functions in gene expression. For example, regulatory functions may involve turning on or off the production of proteins associated with a particular gene, and thereby altering the anatomy or metabolism of an organism. Non-coding DNA sections near the ends of chromosomes help to stabilize the chromosomal structure. Some non-coding sequences are “molecular fossils,” remnants of genes that have been disabled by past mutations. And other sequences appear to be associated with gene variants responsible for inherited susceptibility to some diseases. But still, the biological functions served by the majority of non-coding DNA are not yet understood, despite the fact that much of it seems from comparative genomics studies to be preserved across very many evolutionary generations, either because or in spite of natural selection.
12) DNA Replication and the Nature of Mutations
When living cells with DNA-based chromosomes divide, each daughter cell has to be supplied with a copy of the parent cell’s DNA. The replication process is the basis for biological inheritance. It is aided by the double-stranded structure of DNA and the various steps in the replication process are catalyzed by a variety of enzyme proteins. The basic processes are illustrated in Fig. 12.1. Replication begins by breaking hydrogen bonds between base pairs to “unzip” the two strands of the double helix. Each separated strand then serves as a template for producing a new partner strand with matched base pairs.

As suggested by the green arrows on the partner strands in Fig. 12.1, the synthesis of the partners always proceeds in one direction, while the two strands of the double helix have sugar-phosphate backbones characterized by opposite directionality. The partner synthesis always proceeds from strand ends marked by a phosphate group toward ends marked by a hydroxyl (oxygen bonded to hydrogen) group. The triplet group specifications in the coding table of Fig. 11.3 specifies nucleobase sequences proceeding in that same direction.
The enzyme-catalyzed synthesis of partner strands starts with a short RNA fragment and proceeds to add one matched nucleotide at a time to the template strand from the original DNA molecule. In some cases, as in humans, the catalyzing enzyme has a proofreading capability built in, so that it can remove a newly added nucleotide at the end of a strand being synthesized if it senses that the nucleobases have been accidentally mismatched. The rate at which the partner strand synthesis proceeds has been measured for E. coli to be about 750 nucleotides added per second. When the DNA replication is completed, there is also a repair mechanism that can distinguish base pair mismatches that survived the proofreading during synthesis.
Biochemical details of these replication processes are described elsewhere, but are not essential for our discussion here. The bottom line is that DNA replication is done with a high degree of copying fidelity, but mutations nonetheless occasionally creep in, especially when a template DNA strand has localized damage arising either naturally or through exposure to some external physical (e.g., radiation) or chemical (e.g., carcinogen) agent. The error rate in human DNA replication has been estimated to be about one per ten billion base pairs, which sounds awfully good. But the full human genome, summed over all 46 chromosomes, comprises about three billion base pairs. Thus, there is a roughly 30% probability of getting at least one mutation per cell division. The generation of human sperm generally involves several hundred cell divisions. So something on the order of 100 mutations are passed along to the genome of each new individual. In species with less sophisticated proofreading and post-replication repair mechanisms, the mutation rate can be far greater.
The occurrence of a mutation during a cell division is a random event. But the nature of the possible mutations is not completely random, as the biochemistry establishes some preferences. For example, as indicated in Fig. 11.1, the chemical base structure for adenine is most similar to that of guanine, while that of cytosine is most similar to that for thymine. Correspondingly, the most likely point mutations in DNA replication involve the accidental substitution of A for G, or vice versa, or of C for T, or vice versa. But the probability of occurrence of such a point mutation also depends on the identity of the base’s neighboring bases along the template DNA strand.
Many types of mutation involve more than a single point within the DNA strand. Segments of DNA known as mobile genetic elements (thought to comprise about half of the human genome) may be deleted or inserted during DNA replication, and especially in cell formation of offspring when distinct chromosomes from the two parents are combined. The directionality of a DNA segment can get inadvertently reversed. These deletion, insertion and inversion errors can lead to a variety of large-scale changes to chromosomes, indicated schematically in Fig. 12.2, including duplication or disappearance of genes, and relocation of genes within a single chromosome or even from one chromosome to another. Some of these mutations can arise also not during reproduction, but rather via horizontal gene transfer from a different organism, or even a different species, during cell-to-cell contact in the case of bacteria, or transport by a virus, or genetic engineering in modern biotechnology.

When genes get duplicated, it becomes possible via subsequent mutations for one or more copies of the gene to acquire new functionality, while other copies maintain their original function. This appears, for example, to be the mechanism by which color vision has evolved in vertebrates: a single ancestral light-sensing gene eventually spawned four human eyesight genes, three for cone cells providing color vision and one for rod cells providing night vision. Relocation of a gene can also sometimes lead to new functionality via combination with its new neighbor segments.
In terms of their effects on the fitness of an organism to adapt to its environment, mutations can be harmful, neutral or beneficial. Harmful mutations typically deteriorate or kill the proper functioning of specific proteins, or of regulatory or metabolic networks, that are critical to an organism’s survival or reproduction. They may result in disease or premature death. If the organism can still reproduce successfully, but it passes the harmful mutation along to progeny, this can result in hereditary diseases. Harmful mutations are disfavored by natural selection, so that they tend over many generations to show up with reduced frequency in species populations, or else to get counteracted by subsequent mutations.
Beneficial mutations, though relatively rare, can increase reproductive efficiency or enhance survival probabilities in the ambient environment, or occasionally even introduce innovative biological functionality. These are then likely to be passed on to multiple future generations, where their advantage may sometimes be further enhanced by subsequent mutations. Similarly, a neutral mutation – one with no significant effect on biological functions – introduced in one generation may sometimes combine with other mutations introduced in succeeding generations to form a beneficial change. Indeed, Andreas Wagner and his collaborators at the University of Zurich have shown that the vast library of available silent and neutral mutations, and even some mildly deleterious ones, can provide myriad paths toward critical further mutations that, in subsequent generations, introduce truly innovative adaptations in biological functionality. We will explore this concept in more detail in the next section, because it is critical to understanding evolution.
We also note that certain mutations may be harmful for some purposes but beneficial for others. For example, as we have explained in the biogeography section of part II of this series, human blood cells deformed by the sickle cell mutation are less efficient than normal round blood cells, but are more resistant to malaria. The generational persistence of such a mutation then depends on the environment in which organisms operate.
13) Redundancy, Robustness and Natural Selection Forcing
Creationists and Intelligent Design advocates often claim that there is negligible probability to develop complex life forms by the accumulation of randomly sampled mutations, the vast majority of which they claim are harmful. They sometimes attempt to back up this claim by quoting the astrophysicist Fred Hoyle, who said: “The chance that higher life forms might have emerged in this way is comparable to the chance that a tornado sweeping through a junkyard might assemble a Boeing 747 from the materials therein.” Hoyle was actually discussing his views on the origins, rather than the evolution, of life. But in either case, he and those who quote him make the fundamental mistake of assuming that life relies on finding the “one true path” to biology among an astronomical number of paths for assembling macromolecules. The “junkyard tornado” analogy even goes further by assuming that finding that path has to be done in a single step. ID advocates who accept that there is an accumulation of many steps still argue that there has not been sufficient time on Earth to randomly sample all possible paths to find the right one to each species. The truth is very far from these fictions.
First of all, the majority of mutations are not harmful, but rather are neutral. This results from a high degree of redundancy and robustness against small changes that is built into biomolecules. For example, Andreas Wagner points out that proteins with quite different amino acid sequences perform the same, or very similar, functions – e.g., oxygen transport – in a variety of different organisms. If one thinks of proteins as chains of 100 amino acids, with each acid in the sequence to be chosen from among 20 varieties used in the human genome, then there are 20 to the 100th power, or 10130, conceivable proteins that could be constructed. This is an enormous number. But estimates carried out for some specific proteins suggest that among all these conceivable sequences there can be as many as 1050 or more distinct sequences that all accomplish the same biological function and can be thought of as belonging to a huge genotype network! There are consequently a large number of non-silent point mutations to coding DNA that may alter an amino acid at one location in a specific protein without altering the protein’s function. Such mutations simply transform the protein to another member of the same genotype network.
Starting from any member of such a network, there are also a small number of point mutations that may lead to either harmful or beneficial changes in biological function, by connecting to a different genotype network, which also contains an enormous number of possible amino acid configurations. The consequence of all this redundancy is that there is no “one true path,” but rather very many possible paths that can be found to lead to the same eventual beneficial or harmful mutation, following different sequences of neutral mutations. Evidence for this abounds in the very different amino acid sequences found by different species for certain protein functions, e.g., oxygen transport in hemoglobin in plants vs. insects. As Wagner points out, the same basic story of enormous redundancy and myriad alternative paths to beneficial functioning applies to regulatory gene networks and to the chains of chemical reactions that constitute metabolism.
The redundancy also implies that organisms are remarkably robust against many small changes to their genome. For example, one of the ways bacteria have developed resistance to penicillin is by evolving a protein that cleaves the penicillin molecule, thereby destroying its effectiveness. Researchers have studied how this protein is affected by mutations, and found that 60% of the nearly 1000 point mutations (changes to a single amino acid in the protein molecule) examined had no effect on the penicillin-cleaving function. This robustness extends even to the level of entire genes. In a laboratory at Stanford University, researchers have analyzed the functions of the 6000 or so genes contained in the genome of brewer’s yeast. They did this by knocking out each gene, one at a time, to create 6000 different yeast strains whose subsequent development they could track in the laboratory. They found that deletion of some 40% of the genes had no observable effect whatsoever on the subsequent functioning of the yeast cells.
The redundancy and robustness of these biological functions may well rely on the use of long macromolecules and complex genetic networks, as shorter and simpler ones tend to rely on every one of their components and thus to be more fragile under any sort of disruption. The redundancy and robustness, in turn, enhance the ability of organisms to sample an enormous range of mutations in search of ones that increase fitness in the organism’s environment. There is quite likely a higher probability in these broad-ranging samplings to eventually encounter a harmful mutation than a beneficial one. But the harmful examples tend to die away quickly, and we deal primarily in our observations and research with the highly selected sample of surviving species. And among the beneficial ones, there may occasionally be mutations that produce truly innovative advances in functionality that lead to a branching in the tree of life. We will discuss one particular example in the next section.
There is, then, no reason to assume that the same sort of multigenerational random mutation plus natural selection observed in the laboratory for rapidly reproducing systems (see Part II of this blog) could not have accounted for the bulk of species formation and adaptation over the multibillion year history of life on Earth. It is easy to underestimate the strength of natural selection pressure imposed by survival and multiplication at each stage of an evolutionary path. Richard Dawkins, in his 1986 book The Blind Watchmaker, describes a simple computer code he wrote to illustrate the enormous probabilistic advantages of a multistage approach of random search plus imposed selection, in addressing a problem of much less complexity than life formation and evolution, but one nonetheless facing dauntingly long odds.
The challenge Dawkins set himself was to see if a 28-character string, in which each character was chosen randomly from among 27 possibilities (the 26 letters of the English alphabet plus a space), could reproduce a specific short line from Hamlet: “Methinks it is like a weasel.” There are 2728 or 1.2 × 1040 different possible choices of 28 characters from among 27 options. If one followed the “junkyard tornado” approach of searching for a match to Hamlet in one fell swoop among all these possibilities, the chances are negligible that one would find a match during the lifetime of the universe! A computer capable of checking one random sequence per microsecond could sample about 30 trillion sequences per year but would still require, on average, more than a trillion trillion years to find a perfect match without a selective algorithm to streamline the search. (Long passwords are hard to crack!)
Dawkins instead programmed an iterative approach meant as a crude analog to evolution, in which he chose a purely random 28-character string to start, evaluated random point mutations to each character in the string, and then selected the “fittest” result as the starting point for the next iteration (generation). Fitness was measured by the number of characters in each examined string that matched the target quote in the correct positions. The initial random selection matched three characters out of 28. In the early stages, the vast majority of point mutations sampled were neutral, neither improving nor deteriorating fitness. But occasional mutations were beneficial, increasing the character match by one. The selection of the fittest result at the end of each iteration led to a gradual evolution toward the correct phrase, generally changing only a single character per iteration, but sometimes changing none or two, and occasionally even mutating away from a match in one position to be compensated by a simultaneous beneficial mutation at another.
Dawkins’ code achieved a perfect match to the target phrase in the 43rd generation, after a total of 11 seconds of computer time. His example is certainly an imperfect analog to evolution of the species, in that Dawkins’ criterion for selection was artificial, ruthless and tied to a known “endpoint,” while evolution’s is natural, less draconian in immediately killing off all but the very best descendants, and tied to successful propagation of the species in the absence of a known endpoint. Nonetheless, the reduction from a trillion trillion years of junkyard tornado searching to 11 seconds of iterative searching with selection indicates clearly the importance of a selection criterion and many alternative paths to incremental improvement in reaching an apparently improbable stage efficiently as the cumulative effect of an iterative trial and error process. Redundancy, robustness and natural selection forcing are critical to achieving the evolution of the species during the age of the Earth.
14) Genomic Similarities Among Different Species
Perhaps the strongest evidence supporting the descent of all living creatures on Earth from a common ancestor is the experimental observation that the basic life processes of replication, heritability of characteristics from ancestors to descendants, biochemical catalysis, and energy utilization (metabolism) are carried out by exceedingly similar molecular structures and mechanisms in all studied species. All rely on DNA or RNA polymers assembled from a total of five distinct nitrogenous bases (see Fig. 11.1), chosen among hundreds of such organic compounds that occur naturally or have been artificially synthesized. All known organisms carry out catalysis using enzyme proteins constructed from
22, among the 500 or so known, types of amino acid. So all species on Earth are using the same genetic alphabet. If all human cultures had used a common writing alphabet, it would seem highly improbable that they had developed that alphabet independently. More likely, the alphabet was developed before the various cultures had branched out to diverse destinations.
Even with the restrictions imposed by using the same genetic alphabet, there are over 1070 possible genetic codes to map triplets of the four DNA nucleotides into the 22 amino acids. And yet, all DNA sequencing that has been performed to date indicates that the organisms grouped into the standard tree of life use the genetic code shown in Fig. 11.3, with only slight variations now observed among major taxonomic groups (e.g., vertebrates vs. plants vs. single-celled ciliates). Such commonality was predicted from the theory of universal common descent – and, indeed, was used as a constraint in the cracking of the genetic code in the 1950s and 1960s – because significant changes to the code along an evolutionary path would have affected most of an organism’s proteins and likely led to rapid death. It is a prediction that is confirmed over and over each year as thousands of new DNA and protein sequencing maps are completed.
Comparison of the mapped chromosome structure for different species confirms the basic organization of the tree of life, deduced previously from anatomical similarities. In particular, chromosomal similarities are greatest for species that are most closely related. For example, Fig. 14.1 compares human and chimpanzee chromosomes. The two sets are strikingly similar, with the most glaring difference being a single human chromosome (#2) that appears to be a fusion of two shorter chimpanzee chromosomes (#2A and 2B). The fusion is indicated by the appearance of characteristic chromosome terminal nucleotide sequences (so-called telomeres) not only at the ends, but also near the middle, of human chromosome 2. Nine of the other corresponding chromosomes have some significantly different segments between humans and chimpanzees. These differences often involve genes associated with speech and hearing or immune system defenses, as might be expected from human language development and the evolutionary need for the two species to develop resistance to sometimes different diseases.
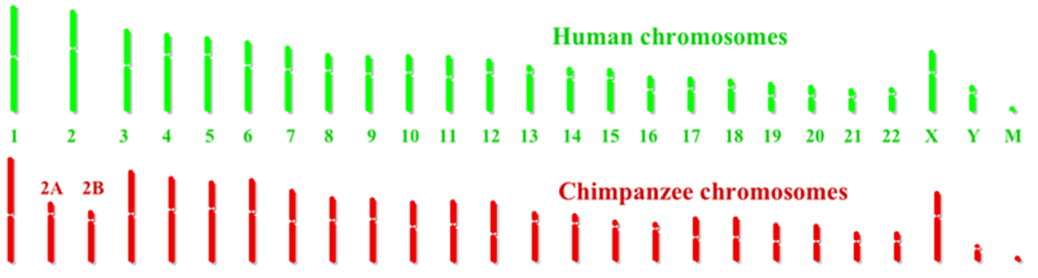
It is conceivable that the fusion of two ape chromosomes to form human chromosome 2 was a triggering event in the separation of the human and chimpanzee evolutionary lineages some 6-8 million years ago. Inserted at the fusion point of the two chromosomes are new genes not present in the apes, one of which corresponds to a bacterial enzyme that makes vitamin B12. B12 is important for brain development, and a major change in human development is greater post-natal brain growth than is observed in other apes. On the other hand, humans seem to have lost, in the evolutionary branching, a functional gene which, in other primates, produces an enzyme that may protect against Alzheimer’s disease.
A detailed comparison between the complete human and complete chimpanzee genomes reveals differences comprising about 35 million single-nucleotide changes, about 5 million mobile genetic element insertion/deletion events, and various rearrangements of genes among chromosomes. Keeping in mind that the human genome comprises some three billion nucleobase pairs, the genomes of the two species differ by only 1—2%, roughly ten times more than the typical differences one finds between pairs of human individuals. Gene duplications account for most of the sequence differences. It has recently become possible to sequence the genomes of now extinct Eurasian contemporaries of homo sapiens, Neanderthals and Denisovans, from recovered bone fragments. For these species, which branched off from modern human development about 500,000 years ago, the genomic differences from modern humans are at the level of several tenths of a percent, perhaps twice as large as the variations among modern humans.
The similarity between chimps and humans even extends to retrovirus insertions in the genomes. Retroviruses, like those that cause AIDS or a form of leukemia, store their own genetic code in DNA and insert copies of their own genome into that of their host. Such insertions, when they occur in sperm or egg cells, are inherited by all descendants. About 1% of the human genome contains molecular remnants of ancestral retrovirus infections, and some of these are shared with apes at identical chromosomal positions, a coincidence exceedingly unlikely to occur without common ancestry. In fact, by mapping which primate species share which retrovirus insertions, one can infer the phylogenetic branch point among primates at which the original infections must have occurred.
Typical human and chimpanzee corresponding proteins differ from one another, on average, by only two amino acids within macromolecules comprising typically hundreds of amino acids. In fact, 29% of all the proteins used for catalysis by chimps and humans have identical amino acid sequences. This includes, for example, cytochrome c, a ubiquitous protein used by all living organisms to transport electrons in fundamental metabolic processes. However, there are some 1093 different possible sequences of the 100 or so amino acid layers in cytochrome c, and all of these would deliver essentially the same biochemical functionality. The sequences are indeed typically different in the cytochrome c in different species. The fact that the sequences are, against all odds, identical for chimpanzees and humans is another strong indication of common ancestry. Their common cytochrome c protein differs by typically about 10 amino acids from those in all other mammals.
The cytochrome c example is illustrative but not at all unique. The strong biomolecular similarities between closely related species would be extraordinarily improbable in the absence of common ancestry, given the enormously broad leeway the biochemistry often allows in the construction of molecules to accomplish basic life functions.
How do Intelligent Design advocates react to the genomic evidence presented in this part of our blog series? Some downplay the extent of human-chimpanzee similarities, saying they tell us nothing about common ancestry. But others claim that the biochemical similarities among species are, in fact, evidence of ID. Stephen Meyer, one of the architects of ID, puts it this way: “DNA functions like a software program. We know from experience that software comes from programmers. We know generally that information—whether inscribed in hieroglyphics, written in a book or encoded in a radio signal—always arises from an intelligent source. So, the discovery of information in the DNA molecule provides strong grounds for inferring that intelligence played a role in the origin of DNA, even if we weren’t there to observe the system coming into existence.” Meyer does not indicate whether he believes that the astronomical radio signals received from pulsars or the cosmic microwave background radiation received from all directions in the sky carry no information (despite informing us greatly about our universe), or whether, alternatively, he is personally acquainted with their intelligent origin.
Thus, many ID advocates claim both the commonality and relative simplicity of genetic alphabets and coding, and the remarkable diversity of complex biological features in different species, as evidence for intelligent design. This observation highlights the failure of ID as a scientific approach: anything can be claimed to indicate design if one doesn’t have to specify the end purpose or the designer, or predict any outcomes before they are observed. How can one falsify such claims? Indeed, some ID proponents go so far as to support the concept of “guided common descent,” accepting the tree of life but claiming that it, too, is simply an indicator of intelligent design. Why, then, do these ID advocates reject the theory of evolution, which is just a “stone’s throw” from guided common descent? Intelligent programmers often use algorithms that rely on iterative random variations, with selection criteria based on some periodically evaluated metric, to find optimal solutions to a problem. Why wouldn’t an intelligent designer of life allow for random mutations and natural selection as an optimal approach – one confirmed by laboratory observations of microevolution – to generating gradual adaptation, with occasional branching, of living species over billions of years to a changing environment? Without such allowance, why would an intelligent designer build retrovirus insertions into the DNA of some closely related species starting at some definable point in the evolutionary process?
ID relies centrally on the concept of irreducible complexity, the claim that at least some biological features are so elegant and complex, and so reliant on multiple components working in unison, that they could not possibly have arisen from cumulative incremental random mutations and natural selection. As we will discuss in Part IV of this blog series, this concept can be, and indeed has been, falsified.
— To be continued in Part IV —
References:
https://en.wikipedia.org/wiki/DNA
https://en.wikipedia.org/wiki/DNA_replication
A. Wochner, J. Attwater, A. Coulson and P. Holliger, Ribozyme-Catalyzed Transcription of an Active Ribozyme, Science 332, 209 (2011) (http://science.sciencemag.org/content/332/6026/209)
T.A. Kunkel, DNA Replication Fidelity, Journal of Biological Chemistry 279, 16895 (2004) (http://www.jbc.org/content/279/17/16895.full)
S.D. McCulloch and T.A. Kunkel, The Fidelity of DNA Synthesis by Eukaryotic Replicative and Translesion Synthesis Polymerases, Cell Research 18, 148 (2008) (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3639319/)
https://en.wikipedia.org/wiki/Amino_acid
https://en.wikipedia.org/wiki/Protein
http://en.wikipedia.org/wiki/DNA_codon_table
https://en.wikipedia.org/wiki/Non-coding_DNA
https://en.wikipedia.org/wiki/Mutation
https://en.wikipedia.org/wiki/Horizontal_gene_transfer
A. Wagner, The Role of Randomness in Darwinian Evolution, Philosophy of Science 79, 95 (2012) (https://www.ieu.uzh.ch/wagner/papers/Wagner_Philosophy_of_Science_2012.pdf)
A. Wagner, Arrival of the Fittest: Solving Evolution’s Greatest Puzzle (Current Publishing, 2014)
(https://www.youtube.com/watch?v=aD4HUGVN6Ko)
H. Jacquier, et al., Capturing the Mutational Landscape of the Beta-Lactamase TEM-1, Proceedings of the National Academy of Sciences U.S.A. 110, 13067 (2013) (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3740883/)
http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html
Richard Dawkins, The Blind Watchmaker (W.W. Norton & Company, 1996) (https://www.amazon.com/Blind-Watchmaker-Evidence-Evolution-Universe/dp/0393351491)
N. Lehman, Molecular Evolution: Please Release Me, Genetic Code, Current Biology 11, R63 (2001) (https://www.sciencedirect.com/science/article/pii/S0960982201000161)
https://en.wikipedia.org/wiki/Human_Genome_Project
https://en.wikipedia.org/wiki/Chimpanzee_genome_project
https://en.wikipedia.org/wiki/Neanderthal_genome_project
http://humanorigins.si.edu/evidence/genetics
http://humanorigins.si.edu/evidence/genetics/ancient-dna-and-neanderthals
E .D. Sverdlov, Retroviruses and Primate Evolution, BioEssays 22, 161 (2000) (https://www.deepdyve.com/lp/wiley/retroviruses-and-primate-evolution-i2q49pY95r)
L. Stone, P.F. Lurquin, and L.L. Cavalli-Sforza, Genes, Culture and Human Evolution: A Synthesis (Blackwell Publishing, 2007), p. 79 (https://www.amazon.com/Genes-Culture-Human-Evolution-Synthesis/dp/1405131667)
https://en.wikipedia.org/wiki/Cytochrome_c
S.E. Vigdor, Signatures of the Artist: The Vital Imperfections that Make Our Universe Habitable (Oxford University Press, 2018) (https://global.oup.com/academic/product/signatures-of-the-artist-9780198814825?cc=us&lang=en&)
https://science.howstuffworks.com/life/evolution/evolution.htm
http://www.talkorigins.org/faqs/comdesc/section4.html
D. Voet and J. Voet, Biochemistry (John Wiley & Sons, 1995)
https://evolutionnews.org/2014/03/does_genome_evi/
S.C. Meyer, Signature in the Cell: DNA and the Evidence for Intelligent Design (https://www.biblearchaeology.org/post/2009/08/19/Signature-in-the-Cell-DNA-and-the-Evidence-for-Intelligent-Design.aspx)
https://evolutionnews.org/2018/11/intelligent-design-and-common-descent/
